
May 12, 2020
Written by: Nitsan Goldstein
Neurodegenerative diseases are a particularly devastating group of illnesses due to their widespread effects on day-to-day life and the fact that they are extremely difficult to treat. Among the most debilitating of these diseases is Huntington’s disease (HD). HD is almost always inherited from a parent, making it unique among other neurodegenerative diseases that are often sporadic (not directly inherited) and/or arise from a combination of genetic and environmental factors. The disease results from a mutation in a gene called huntingtin. This tiny alteration in one region of our DNA has widespread effects on the brain that result in a host of symptoms for people who have HD. Here, we will discuss the neural circuits that are not functioning properly in HD and how scientists are trying to use what we know to develop treatments.
What is HD?
Huntington’s disease symptoms can develop at any time in a person’s life, but typically begin in middle age. The first symptoms are usually slight changes in mood and behavior along with sudden, uncontrolled movements. Many of these symptoms progressively worsen and patients die 15-20 years after the onset of symptoms1. The progression in motor symptoms, however, is quite distinct. In contrast to the excess movements in the early stages of the disease, the late stages are marked by the inability to initiate movements along with muscle rigidity1. The complexity and unique progression of these motor symptoms is a consequence of disruption to the delicate balance of excitatory and inhibitory signals in our brain that allow us to execute fine movements in a region called the basal ganglia.
A Healthy Basal Ganglia
A primary function of the basal ganglia is coordinating conscious, goal-directed movements. Of course, the brain needs to activate regions that directly control our muscles. However, in order to perform precise movements like picking up a glass of water, our brains also need to inhibit motion that would interfere with this task, like movement of the arm side to side. The brain also needs to prevent movement when the body is at rest and no movement is needed. The intricate circuits in the basal ganglia send both a “move” and “don’t move” signal, and the right balance between these two signals is critical for proper control of movement.
What are these “move” and “don’t move” signals and how do they arise? Neural activity comes into the basal ganglia from two main regions: the cortex and the substantia nigra. Within the basal ganglia, this information can travel either along the direct pathway or the indirect pathway, depending on what kinds of receptors are present on the basal ganglia neurons. Information coming out of the basal ganglia travels to a region called the thalamus and eventually reaches the motor cortex, which directly controls muscles. The direct pathway is the “move” signal and leads to excitation of the motor cortex, while the indirect pathway is the “don’t move” signal and leads to inhibition of the motor cortex (see Figure 1). Basal ganglia neurons and their inputs are particularly affected in HD, leading to the hallmark motor symptoms2. What happens to the basal ganglia and how might it explain such symptoms?
What Goes Wrong in HD?
The first region in the brain known to be impacted in HD is the cortex. Years of work using rodent models of HD suggests that the neurons in the cortex start to become hyperactive2. The primary chemical used by these neurons to communicate with other neurons is glutamate. Glutamate is the primary excitatory signal in the brain, but too much glutamate is toxic to neurons. Remember that the cortex is one of the main inputs to the basal ganglia and is critical in initiating movements. The increased glutamate released from the cortex on the basal ganglia neurons shifts the balance of “move” and “don’t move” signals towards “move”. The neurons in the direct, “move” pathway become hyperactive while the neurons in the indirect, “don’t move” pathway begin to deteriorate. Thus, patients experience uncontrolled movements. As the disease progresses, the increased glutamate coming from the cortex becomes toxic and the neurons of both the direct and indirect pathways degenerate. Eventually, the neurons in the cortex also die, and thus the signal into the basal ganglia to initiate movements stops (see Figure 1). When this happens, patients start to experience muscle rigidity and completely lose the ability to move.
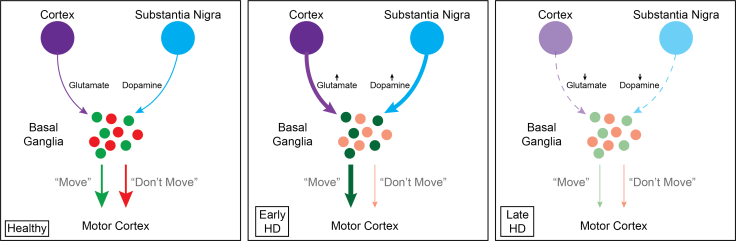
The second input to the basal ganglia, the substantia nigra, is also impacted in HD2. The neurons in the substantia nigra release dopamine in the basal ganglia. Dopamine alters the activity of basal ganglia neurons and, along with glutamate from the cortex, is critical in initiating proper movements and inhibiting unwanted movements. Much like cortical neurons, the activity of these neurons is heightened in the early stages of HD. The resulting increase in dopamine release in the basal ganglia further shifts the signal towards “move” by increasing direct pathway activity and decreasing indirect pathway activity. Eventually, these neurons also begin to degenerate, contributing to the loss of input into the basal ganglia and an overall decrease in motor control later in the disease progression. One study examined the brains of HD patients who had died and found 40% fewer neurons in this region compared with controls, which highlights just how severe the cell loss in this circuit is in the late stages of the disease3.
Treatments
Despite the identification of the mutation in the huntingtin gene that causes HD more than 25 years ago, the treatment options for the disease remain fairly limited4. The early motor symptoms can be treated with a drug called tetrabenazine5. This drug prevents neurons from releasing dopamine, which reduces direct pathway activation and shifts the balance of the basal ganglia signal back away from the excessive “move” signal it sends during this early stage of the disease. Tetrabenazine, however, also prevents neurons from releasing other chemicals such as serotonin. The loss of serotonin signaling can worsen depression and anxiety, which are frequently experienced early on in the disease. Finding the right dose, therefore, is critical for preventing harmful side effects. Anti-psychotic medications such as haloperidol are also frequently used to treat early motor symptoms and, unlike tetrabenazine, can also improve psychiatric symptoms5. These treatments, while important for improving patient quality of life in the early stages of the disease, do not slow the progression of neurodegeneration. The ultimate goal of doctors and researchers is to find a way to delay or stop disease progression at the onset by targeting the root cause of the abnormal neural activity, rather than making symptoms more manageable.
Exciting Future Directions
One of the reasons treatments for HD focus on symptom control and not the cause of the disease itself is because it has been particularly challenging to identify exactly how the mutation in the huntingtin gene results in the widespread neural damage described here. While the precise function of the protein is not clear, studies have identified many key cellular processes that are abnormal when the huntingtin gene is mutated. It is likely a combination of many of these dysfunctional processes that causes hyperactive cortical neurons and the resulting cell death, which makes targeting these processes for treatment difficult5. The best hope for delaying or even curing the disease is in gene therapy (click here for an article on gene therapy in brain damage). Patients with HD have one copy of the normal huntingtin gene and one that is mutated. The protein that forms from the mutated copy is what causes cell damage and eventual death. If that copy of the gene could be silenced, meaning the protein forms from the normal copy and not the mutated one, the disease itself could be slowed or even stopped in its tracks. One common technique involves introducing RNA that precisely targets the RNA produced by the mutant gene so that it is destroyed before it is used to make the dysfunctional protein. Two drugs based on this RNA technology have already been shown to be effective in reducing the levels of the mutated huntingtin protein in the brains of mice and primates with the HD mutation6. These drugs are currently in the early stages of human clinical trials, with very promising results. The hope is that the drugs will prove safe and effective in dramatically slowing disease progression to prolong the lives of HD patients. Thus, while HD is a devastating disease with very little treatment options at the moment, it is a contender for being the first neurodegenerative disease to ever have a cure.
References:
- Walker, F.O. Huntington’s disease. Lancet 369, 218-28 (2007).
- Dudanova, I., Blumenstock, S. Cortical and Striatal Circuits in Huntington’s Disease. Front. Neurosci. 14, 82 (2020).
- Oyanagi, K., Takeda, S., Takahashi, H., Ohama, E., & Ikuta, F. A quantitative investigation of the substantia nigra in Huntington’s disease. Ann. Neurol. 1, 13-19 (1989).
- MacDonald, M.E., et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971-83 (1993).
- Wyant, K.J., Riddler, A.J., & Dayalu, P. Huntington’s Disease—Updates on Treatments. Curr. Neurol. Neurosci. Rep. 17, 33 (2017).
- Leavitt, B.R, & Tabrizi, S.J. Antisense oligonucleotides for neurodegeneration. Science 367, 1428-29 (2020).
Cover Image by Enro2002 and Wikimedia Commons, CC BY-SA 3.0 https://commons.wikimedia.org/wiki/File:Pkan-basal-ganglia-MRI.JPG

{kind=link}