
August 7, 2018
Written by: Katerina Placek
Alzheimer’s disease is the main cause of dementia among older adults, leading to irreversible destruction of a person’s memory and thinking skills and, ultimately, death. While the cause of Alzheimer’s disease remains unknown, the prevailing focus of research has been on the harmful role of a protein called beta-amyloid (Aβ) that is found in the brains of individuals with Alzheimer’s disease and is believed to contribute to neuronal degeneration in regions of the brain important for memory and thinking. However, new research suggests that the brain’s immune system may influence Aβ and play an integral role in Alzheimer’s disease.
What is Aβ?
Before we go any further, let’s first define what exactly Aβ is. Aβ is a product of the β-amyloid precursor protein (APP). Scientists are still characterizing the function of APP, but believe it is used in the wall-like membranes that surround neurons and in the synapses that allow different neurons to communicate with one another. APP is then processed by enzymes, which ‘fine tune’ the protein for usage by the neuron. When APP is processed in a specific manner, Aβ is produced and immediately released outside the neuron. Like APP, the function of Aβ is not well understood. However, when Aβ accumulates outside the neuron, it can clump together and form sticky deposits called plaques (yes, like dental plaque) that are found in the brains of individuals with Alzheimer’s disease.
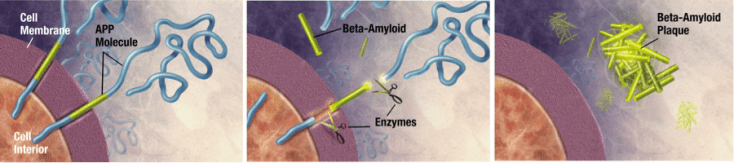
Does Aβ cause Alzheimer’s disease?
Until recently, the prevailing idea among Alzheimer’s disease researchers was that the accumulation of Aβ into plaques initiated a cascade of events leading to degeneration of neurons and, ultimately, the devastating cognitive decline and memory loss associated with Alzheimer’s disease dementia.
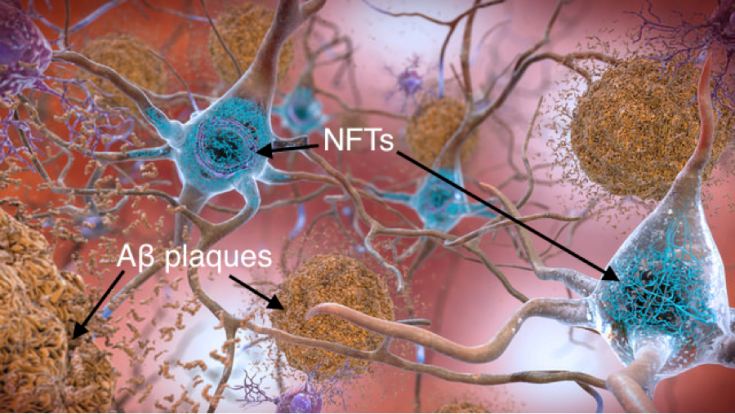
For proponents of this idea, the formation of Aβ plaques outside the neuron triggers the formation of neurofibrillary tangles (NFTs) inside the neuron, 1 as pictured in Figure 2. NFTs are important because Aβ plaques and tau NFTs are the two “hallmarks” of Alzheimer’s disease required for diagnosis. NFTs and Aβ plaques together are then believed to lead to dysfunction of the neuron and its synapses, and finally neurodegeneration.
The initiating role of Aβ in this cascade of events, along with genetic evidence of APP mutations in patients with Alzheimer’s disease, has been interpreted as evidence for the primary pathologic role of Aβ.2 Accordingly, therapeutic targets for the treatment of Alzheimer’s disease have largely focused on preventing Aβ formation or clearing Aβ.3
Immune response – consequence, contributor, or protector?
In addition to Aβ plaques and tau NFTs, Alzheimer’s disease also features an inflammatory response driven by the brain’s immune system.
This inflammatory response features the increased activity of microglia and astrocytes, which are both types of supportive cells in the brain and spinal cord. Microglia function as the first and main form of active immune defense, constantly scavenging for plaques, damaged neurons and synapses, and infectious agents to digest and destroy. Astrocytes are the most numerous cell type in the brain and support the blood-brain barrier, provide nutrients, and repair tissue in response to injury.
According to researchers who think Aβ is the harmful initiator of Alzheimer’s disease, this inflammatory response is a consequence of Aβ plaque formation.2 In this view, microglia activate to degrade Aβ plaques and clear dysfunctioning/degenerating neurons, and astrocytes activate to support and repair damaged neurons. Support for this interpretation comes from the observation that microglial and astrocyte activity increase with Aβ plaque formation and neuronal dysfunction/degeneration.
However, new research suggests that the inflammatory response is not merely a consequence of Alzheimer’s disease pathology but may actively contribute to disease proliferation. A recent study compared the DNA of Alzheimer’s disease patients relative to healthy people, and discovered that Alzheimer’s disease patients featured mutations in genes important to microglia that the healthy people did not.4 This suggested that the immune response of microglia contributes directly to the development of Alzheimer’s disease, rather than acting only consequentially to pathologic accumulation and neurodegeneration. In further support of this idea, researchers have also found that activated astrocytes actually produce Aβ in response to pro-inflammatory signals, and thus contribute to the overall Aβ burden in the brain!5
Last, and most shockingly, some scientists think that Aβ is part of the brain’s innate immune system, and normally plays a protective role – except when produced in excess, which leads to Alzheimer’s disease. In support of this idea, Aβ has been shown to have anti-microbial (germ-killing) properties in multiple species.6 Interestingly, this raises the possibility that infectious or inflammatory stimuli can drive over-production of the normally protective Aβ, and suggests a dual protective/damaging role for Aβ. Scientists are now actively researching how this immune ‘glitch’ may lead to Aβ overproduction of a-beta.
Why does this matter?
The idea that the immune system may contribute to Alzheimer’s disease holds exciting implications. In particular, scientists may focus on developing therapeutics that target the brain’s innate immune system, or triggers of the innate immune system, rather than Aβ itself. This may lead to better treatments for patients suffering from Alzheimer’s disease, and a better understanding of how Alzheimer’s is caused – and, hopefully, how to prevent it.
References:
- Choi, S. H. et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278 (2014).
- Hardy, J. & Selkoe, D. J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 297, 353–356 (2002).
- Baranello, R. J. et al. Amyloid-beta protein clearance and degradation (ABCD) pathways and their role in Alzheimer’s disease. Curr Alzheimer Res 12, 32–46 (2015).
- Sims, R. et al. Rare coding variants in <i>PLCG2</i>, <i>ABI3</i>, and <i>TREM2</i> implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 49, 1373–1384 (2017).
- Zhao, J., O’Connor, T. & Vassar, R. The contribution of activated astrocytes to Aβ production: implications for Alzheimer’s disease pathogenesis. J Neuroinflammation 8, 150 (2011).
- Soscia, S. J. et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS ONE 5, e9505 (2010).
Image References:
Cover image: Photo by Cheron James on Unsplash
Figure 1: By derivative work: Garrondo (talk)Amyloid_01big1.jpg: ADEAR: “Alzheimer’s Disease Education and Referral Center, a service of the National Institute on Aging.” [Public domain], via Wikimedia Commons
Figure 2: National Institute of Aging, NIH; Attribution-NonCommercial 2.0 Generic (CC BY-NC 2.0) https://www.nia.nih.gov/health/what-happens-brain-alzheimers-disease
