September 10, 2019
Written by: Claudia Lopez-Lloreda
“The index case for the present study was a ten-year-old child, well known to the medical service after regularly performing ‘street theatre’. He placed knives through his arms and walked on burning coals, but experienced no pain1.” This unique case of a Pakistani boy led to an important study that unraveled key mechanisms of feeling pain. Studying three different families in Pakistan, a group of researchers found six individuals that had never experienced pain1. They could feel temperature, tickling, and slight pressure, but not pain. All had injuries to their lips and/or tongue caused by biting themselves, and had frequent bruises and cuts. Amazingly, however, they related how they had never felt pain in any part of their body at any point in their lives.
Although most of us think we would be better off not feeling pain, this condition, known as congenital indifference or insensitivity to pain (CIP), can actually be very dangerous. Despite it being extremely rare (about 20 cases have been reported in the scientific literature), those that suffer from it tend to experience a more than average amount of fractures, bruises, and cuts due to their inability to perceive painful stimuli. Therefore, understanding what is happening in the central nervous system of these patients could help develop therapies for them as well as help understand the mechanisms that lead people to feel pain normally.
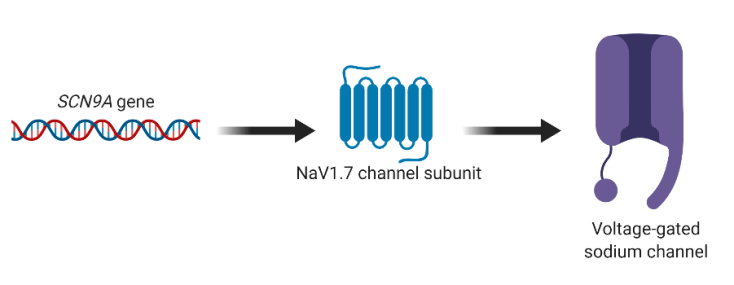
Delving into analyzing these individuals, scientists found the answers within their genes: by sequencing their DNA, researchers found that gene mutations were associated with the condition. The mutations were autosomal recessive, meaning that two copies of the mutations (one from the mother and one from the father) must be present for CIP to develop. Specifically, the mutations were in the SCN9A gene, which encodes for NaV1.7, a subunit of a protein called a voltage-gated sodium channel (Figure 1). These channels are usually expressed in cells called nociceptive neurons that are located throughout the body and sense pain. More importantly, the function of the channel subunits is to help and facilitate the conduction of action potentials, the electrical basis of communication in neurons. This finding hinted at the idea that somehow these mutations affected neuronal communication in pain-sensing neurons of afflicted patients.
One way a mutation could impact a protein is by causing a change that leads to less or no function of the protein. In fact, loss-of-function of NaV1.7 has been studied before in rodents. Mice that do not have NaV1.7 in nociceptive neurons have severely affected ability to feel pain, including inflammatory pain2. Not surprisingly, when researchers examined the consequences of the patient mutations, they found that they altered the structure of the protein, leading to “broken” NaV1.7 ion channels that could not perform their normal functions1. Subsequently, more studies have looked at other patients and found more mutations in the SCN9A gene that are associated with partial insensitivity to pain3, further pointing to this gene as very important in controlling pain.
Interestingly, researchers also found a role for NaV1.7 ion channel subunits on the other side of the balance: people that feel too much pain. Studies found that mutations in SCN9A were also present in patients with primary erythromelalgia, a condition characterized by attacks of burning pain of the feet and lower legs 3. However, in contrast to the mutations associated with CIP that were loss-of-function, these mutations were gain-of-function mutations, those which lead to enhanced activity of a protein (Figure 2). For example, one mutation led to the slower deactivation of the channel, meaning it was open for a longer amount of time and by consequence more “active”4. Another mutation had a clear functional effect on activity: neurons fired more action potentials5. These studies indicated that losing NaV1.7 function leads to an inability to feel pain while overactivation of NaV1.7 function leads to more pain. Therefore, dysfunction of this ion channel subunit seems to be a critical point that controls how much or how little pain we feel.

Pharmaceutical industries have noticed the potential of these findings. Even though CIP patients seem to be a reminder that not feeling pain could be detrimental, researchers and pharmaceutical industries have sought out to target and inhibit NaV1.7 as a way to control chronic pain, especially as an alternative to opioid pain-killers6. Many NaV1.7 inhibitors have been or are in the discovery or clinical trials phases, but the trek has been difficult. As there are other voltage gated sodium channels and other NaV subunits, drugs have to be incredibly specific to NaV1.7 to be able to have the desired effect. Nonetheless, this story highlights how pathological conditions could lead us to understand commonplace mechanism and how one tiny little protein could be a big player in pain.
References
- Cox, J. J., Reimann, F., Nicholas, A. K., Thornton, G., Roberts, E., Springell, K., … Woods, C. G. (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature, 444(7121), 894–898. doi: 10.1038/nature05413.
- Nassar, M. A., Stirling, L. C., Forlani, G., Baker, M. D., Matthews, E. A., Dickenson, A. H., & Wood, J. N. (2004). Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proceedings of the National Academy of Sciences, 101(34), 12706–12711. doi: 10.1073/pnas.0404915101.
- Yang, Y., Wang, Y., Li, S., Xu, Z., Li, H., Ma, L., … Shen, Y. (2004). Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. Journal of Medical Genetics, 41(3), 171–174. doi: 10.1136/jmg.2003.012153.
- Tanaka, B. S., Nguyen, P. T., Zhou, E. Y., Yang, Y., Yarov-Yarovoy, V., Dib-Hajj, S. D., & Waxman, S. G. (2017). Gain-of-function mutation of a voltage-gated sodium channel NaV1.7 associated with peripheral pain and impaired limb development. Journal of Biological Chemistry, 292(22), 9262–9272. doi: 10.1074/jbc.m117.778779.
- Dib-Hajj, S. D., Rush, A. M., Cummins, T. R., Hisama, F. M., Novella, S., Tyrrell, L., … Waxman, S. G. (2005). Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain, 128(8), 1847–1854. doi: 10.1093/brain/awh514.
- Kingwell, K. (2019). Nav1.7 withholds its pain potential. Nature Reviews Drug Discovery. doi: 10.1038/d41573-019-00065-0.
Images
Cover image from Esther Max on Flickr, CC BY 2.0.
Figure 1. Made with BioRender.
Figure 2. Made with BioRender.

Leave a comment